I denne posten tenkte jeg å fortelle litt om det som skjer på området for merking av medisinsk og medisinteknisk utstyr i USA og litt om hvordan dette utvikler seg i Europa. Per i dag så er det USA som har kommet lengst i regulering av dette området, men EU er også i ferd med å utarbeide og vedta direktiv som vil tilsvare lovgivningen i USA på området. Planene for merking av medisinsk utstyr, og etablering av sentraliserte databaser med informasjon om medisinsk utstyr, vil gi store fordeler for alle aktører som har befatning med slikt utstyr.
UDI i USA
I 2013 vedtok US Food and Drug Administration, FDA, å etablere et system for unik identifisering som er tilrettelagt for å kunne identifisere medisinsk utstyr fra fabrikk til bruker. Reglene krever at de som er «ansvarlig for å merke utstyr» skal merke utstyr og utstyrets forpakning med en unik utstyrs-identifikator, UDI, bortsett fra noen unntakstilfeller som er fastsatt i regelverket.
En UDI skal oppgis både i en form som er lesbar for mennesker og i en form som støtter teknologi for automatisk identifikasjon og datafangst (AIDC).
Det er også krav om at UDI skal være merket på selve det fysiske utstyret dersom utstyret er beregnet for mer enn én gangs bruk der utstyret må gjennomgå en behandling, eksempelvis sterilisering, før hver bruk.
Videre så skal datoer på merking av utstyr og forpakninger presenteres på et standard format som er konsistent med internasjonale standarder og internasjonal praksis.
En UDI er i følge FDA en unik numerisk eller alfanumerisk kode som består av to deler:
- En utstyrsidentifikator, UI (engelsk: device identifier, DI), som er obligatorisk. Den skal utgjøre en fast andel i antall posisjoner av den totale UDI, og skal identifisere ansvarlig merker (aktør som er ansvarlig for merking), og identifisere spesifikk versjon eller modell av utstyret.
- En produksjonsidentifikator, PI, som er en betinget, variabel andel av UDI, og som identifiserer en eller flere av følgende attributter:
- Lot eller batchnummer som en instans av et utstyr er tilordnet i produksjon av utstyret.
- Serienummer for en instans av et utstyr.
- Utløpsdato for en instans av et utstyr.
- Produksjonsdato for en instans av et utstyr.
- Den spesifikke identifikasjonskoden som er påkrevd ved US lov §1271.290(c) for menneskelige celler, vev, eller cellulære og vevs-baserte produkt (HCT/P) som er regulert som medisinsk utstyr.

Alle UDIer skal utstedes under et system som forvaltes av en FDA-akkreditert utstedervirksomhet. Regelverket fra FDA beskriver en prosess for hvordan en virksomhet kan søke om akkreditering, hvilke informasjon som søker må oppgi, og hvilke kriteria som vil legges til grunn ved vurdering av slike søknader.
Videre så er visse unntak og alternativ beskrevet i regelverket for å sikre at kostnader og ulemper holdes på et så lavt nivå som mulig.
UDI systemet vil bli implementert i faser over en periode på syv år for å sikre en smidig implementering og for å fordele kostnader og byrder over tid.
Som en del av systemet er de som ansvarlig for merking pålagt å levere informasjon til den FDA administrerte «Global Unique Device Identification Database», GUDID. GUDID vil inkludere et standard sett med grunnleggende identifikasjons-elementer for hvert utstyr som har en UDI, og inneholder kun informasjon om UI (DI). Det er UI som vil være nøkkelen for å finne informasjon om utstyret i databasen. PI (produksjonsidentifikatorer) er ikke en del av GUDID. Mesteparten av informasjonen vil gjøres offentlig slik at brukere av medisinsk utstyr enkelt kan finne informasjon om et utstyr. En UDI indikerer ikke, og GUDID databasen inneholder ikke, noe informasjon om aktører som bruker et utstyr.
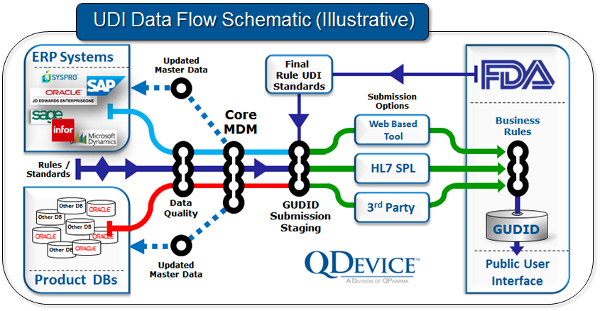
For mer informasjon om GUDID og UDI kan man gå til UDI ressurs sidene hos FDA der det er både opplæringsmateriell, veiledninger og annet UDI-relatert materiale. British Standards (BSI) har også publisert et dokument om UDI.
Fordeler med et UDI system
Når systemet er fullt ut implementert så vil UDI-systemet føre til en rekke fordeler for leverandørindustrien, FDA, forbrukere og helseforetak gjennom å:
- Legge til rette for mer presis rapportering, tilsyn og analyse av avvik slik at problem-utstyr kan identifiseres og korrigeres raskt og effektivt.
- Redusere medisinske feil ved at helsearbeidere og andre raskere og mer presist kan finne viktig informasjon om et utstyr og dets egenskaper.
- Tilrettelegge for analyser av utstyr på markedet gjennom å tilby en standardisert tilnærming for å dokumentere bruk av utstyr i kliniske systemer, pasientjournaler og refusjonsprosesser.
- Tilby en standardisert identifikasjon som vil gjøre det mer effektivt for produsenter, distributører og helseforetak å håndtere tilbakekalling av medisinsk utstyr fra markedet.
- Tilrettelegge et grunnlag for en global og sikker distribusjonskjede, og der igjennom, blant annet redusere problemet med forfalskninger.
- Utvikling av et globalt anerkjent system for identifisering av medisinsk utstyr.
Når kommer dette til Europa?
EU Kommisjonen publiserte sine anbefalinger for et direktiv angående et rammeverk for unik identifisering av medisinsk utstyr 5. april 2013. Man har antatt at det vil ta to til tre år før dette kommer i lovs form, og man kan dermed forvente at dette vil komme tidligst i slutten av 2015 eller i løpet av 2016.
EU er oppmerksom på at regulering på dette området bør harmoniseres globalt da det vil være ineffektivt for leverandørindustrien dersom det stilles ulike krav på dette området i de store handelsområdene. I EU er man bekymret for at det skal etableres nasjonale databaser som følger en avvikende praksis før nytt regelverk og en europeisk informasjonsdatabase er på plass.
Noen begrepsforklaringer
- Ansvarlig merker – En som er ansvarlig for å merke et utstyr er den aktøren som besørger at et utstyr blir merket, eller som besørger at en merking av et utstyr blir modifisert med den hensikt å kommersielt distribuere utstyret uten dertil etterfølgende erstatning eller modifisering av merkingen. Tillegg av navn på, og kontaktinformasjon for, en aktør som distribuerer utstyret uten at det gjøres andre endringer av merkingen er ikke å oppfatte som en modifisering som berører hvem som skal ansees som ansvarlig merker av utstyret. I de fleste tilfeller så vil ansvarlig merker være fabrikanten av utstyret, men ansvarlig merker kan også være de som er ansvarlig for design av utstyret, de som re-prosesserer brukt utstyr og de som setter sammen utstyrssett.
- Automatisk identifikasjon og datafangst, AIDC – Dette innebærer enhver teknologi som kan formidle UDI identifikatorer av utstyr på en slik måte at de kan registreres i et system med pasientinformasjon eller andre informasjonssystemer.
